FDA grants priority review to argenx sBLA for seronegative myasthenia gravis
argenx says the FDA accepted a supplemental biologics license application for IV VYVGART in AChR‑Ab seronegative gMG with a PDUFA date of May 10, 2026; CIDP filing was previously fast-tracked.

The U.S. Food and Drug Administration has accepted for priority review a supplemental Biologics License Application from argenx SE for intravenous VYVGART (efgartigimod alfa-fcab) to treat adults with acetylcholine receptor antibody (AChR‑Ab) seronegative generalized myasthenia gravis (gMG). The company announced the agency set a Prescription Drug User Fee Act target action date of May 10, 2026.
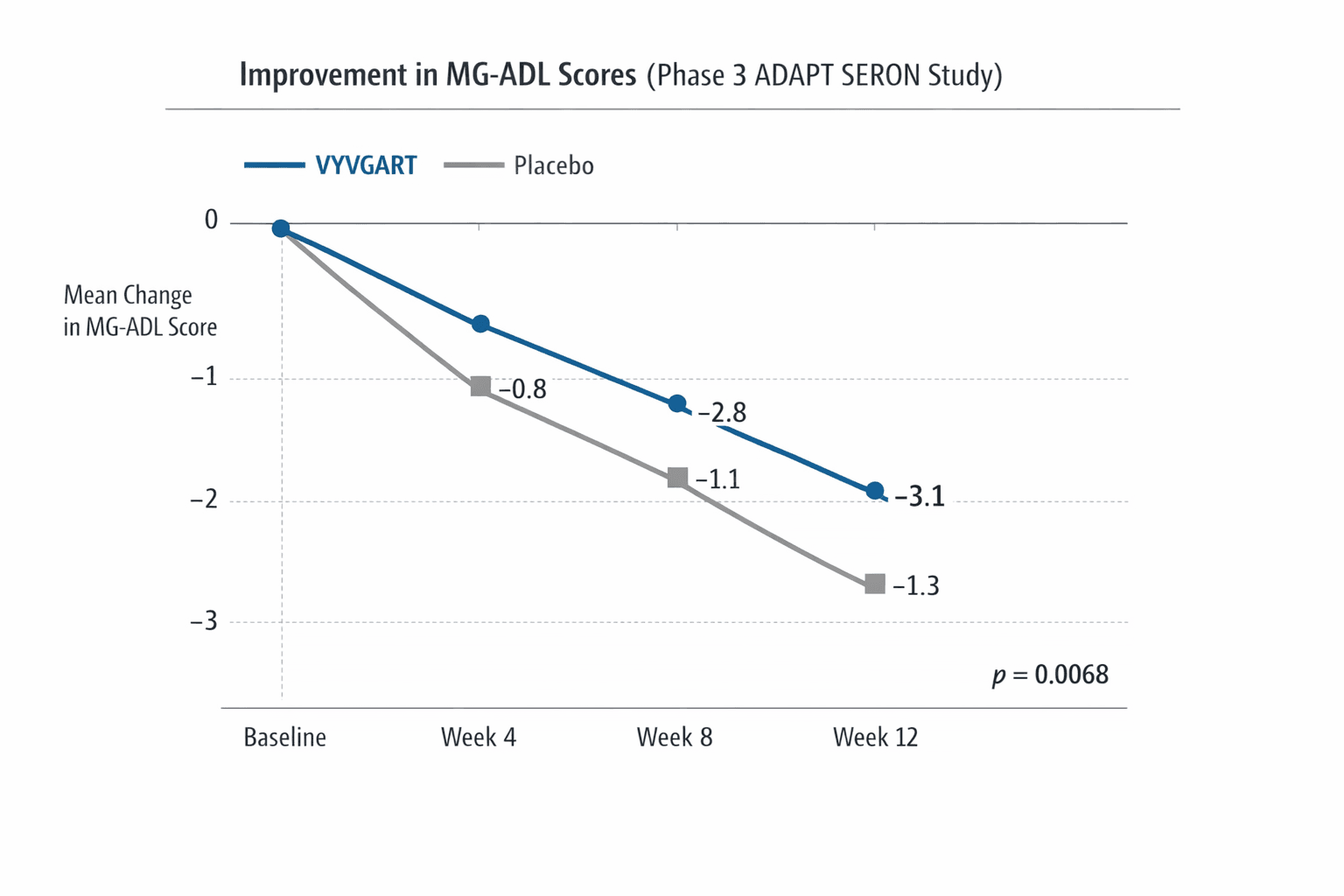
argenx said the sBLA is supported by data from the Phase 3 ADAPT SERON study, which evaluated VYVGART across seronegative subtypes including MuSK‑positive, LRP4‑positive and triple seronegative gMG. The company reported that ADAPT SERON met its primary endpoint, showing a statistically significant improvement in the Myasthenia Gravis Activities of Daily Living (MG‑ADL) total score compared with placebo after four weeks, with a p‑value of 0.0068.
The acceptance follows a separate regulatory path for a subcutaneous formulation, VYVGART Hytrulo (efgartigimod alfa plus hyaluronidase‑qvfc), which argenx previously had accepted for priority review for chronic inflammatory demyelinating polyneuropathy (CIDP) with a PDUFA target action date of June 21, 2024. Company statements say the CIDP filing was accelerated through the use of a priority review voucher; ADHERE, the Phase 3 study cited for that submission, reported a 61 percent lower risk of relapse on active treatment versus placebo and high continuation into an open‑label extension (226 of 228 eligible patients, 99 percent).
argenx’s chief medical officer, Luc Truyen, M.D., Ph.D., framed the FDA action as a response to an unmet clinical need. “Patients living with seronegative gMG continue to face limited treatment options and there remains a significant need to meaningfully improve their lives,” he said in the company release. “The FDA’s acceptance of our sBLA with Priority Review status reflects the potential of VYVGART to address this need. This development brings us closer to expanding the use of VYVGART in a broad spectrum of patients with myasthenia gravis. We look forward to continuing our dialogue with the FDA as they review our application.”
The filings mark an important regulatory moment for rare autoimmune neuromuscular diseases. Seronegative gMG patients historically have been underrepresented in clinical development and have fewer approved options than patients with detectable AChR antibodies. If regulators find the ADAPT SERON evidence persuasive, intravenous VYVGART would extend an FcRn‑directed therapy to a group clinicians have struggled to treat reliably.
Public health and equity questions will follow any approval. Novel biologics for rare diseases tend to be costly and can strain insurance systems and patient finances, raising access concerns for low‑income, rural and underinsured patients. Ensuring equitable access will require proactive payer coverage policies, manufacturer assistance programs and attention to diagnostic equity: serostatus testing and specialty referrals must be available across diverse communities so eligible patients are identified.
Clinicians and patient groups will be watching the FDA timetable and any advisory committee activity as the May 10, 2026 decision date approaches. For CIDP, the earlier priority review and ADHERE results signal continued momentum for argenx’s FcRn platform, but both efficacy in controlled trials and long‑term real‑world safety will determine how broadly these therapies reshape care for people with rare neuromuscular disorders.
Sources:
Know something we missed? Have a correction or additional information?
Submit a Tip